

In the last article I ended having a DNA template thanks to Prof. Yu Fu and I had a DNA sequence of the region where I want to integrate my DNA construct. So to now I need to copy a piece of the DNA plasmid (ring) that I have and add a stretch of DNA sequence (~ 50 bases) that are complementary to to the point in the yeast genome where I want to integrate that peace. I can do both in one step with a technique called Polymerase Chain Reaction or PCR.
A PCR is a method to produce DNA without using living cells to do it. It is something like a molecular copy machine. The main ingredients for a PCR are an enzyme called DNA-polymerase, which does the copying, dNTPs, which are the building blocks of DNA, the template DNA from which I want to copy and primers. Primers are small DNA peaces (normally about 20-25 bases long) that define the DNA stretch I want to copy. The forward primer (the start signal) is complementary to the sequence where I want to start the copy and the reverse primer is identical to the sequence where I want to stop the copy.
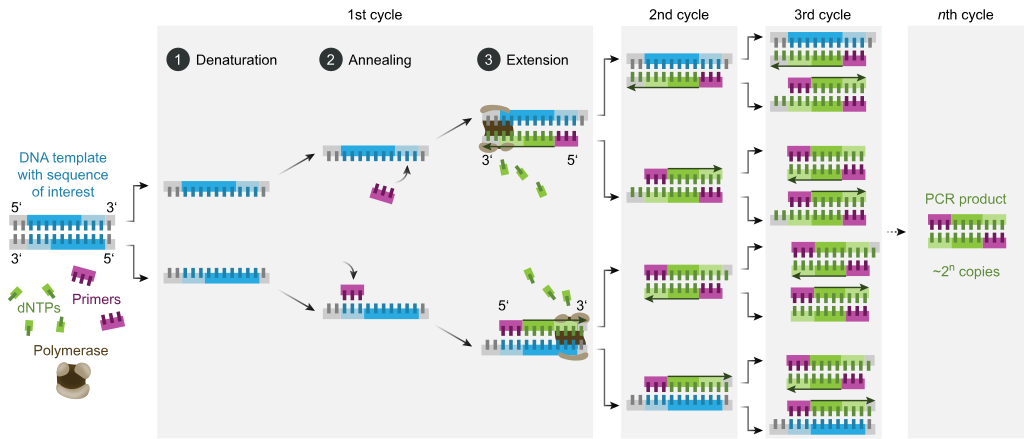
The picture above is from the Wikipedia article on PCR that also explains it really well in detail. As you see the first step of a PCR is making the DNA accessible for the copying by heating it until the typical double-strand structure splits up into two single strands. For this you heat it to something around 95 °C. Then you cool it down to the annealing temperature which depends on your primers but is normally a bit above 50 °C. To start the DNA-polymerase you now heat it again to something around 72 °C. This sounds like a very high temperature for an enzyme and it is. The trick is that the polymerase comes from the bacterium Thermophilus Aquaticus (or others) that lives around hydrothermal vents that spew water, smoke, acid and other stuff at temperatures of more than 100 °C. So their enzymes are optimized to work in extremely hot and unpleasant conditions.
Now you repeat these three steps over and over again and with each repeat the number of your DNA constructs doubles. This means you have an exponential growth from 1 to 2 to 4 to 8… to a bit more than a million after 20 cycles to a bit more than a billion after 30 cycles. So you create a lot of identical DNA in quite a short time (normally 1 to 4 hours).
The machine that adjusts the temperature in a programmed manner is called a Thermocycler and looks quite unspectacular.

This explains how I copy the stretch of DNA from the plasmid but I also wanted to add some sequence to the ends. That’s actually quite easy. I just add the sequence to the primers because they are the beginning and the end of my newly created DNA constructs. Now you might ask: And how do you get these primers? Well like most things today. I order them online. There are companies specialized in producing custom DNA. Basically I could also save myself the whole work and just order my whole construct online but that is still quite expensive. While my exceptionally long primers (primer + sequence are about 60 bases long) cost something between 20 and 50 $, my whole 2500 bases long construct would cost about 1.000 $ to order. That is still quite a difference but it gets cheaper and in a few years biologists will probably order a lot more DNA than they create using molecular techniques like PCR and cloning.
Now that I have my PCR product I want to verify if it is the right one. The easiest way to do that is to put it on a gel that lets short DNA pass quicker than long DNA. Than you add voltage and the negatively charged DNA wanders towards the positive electrode and splits up by size. If you add a marker that includes DNA pieces of known size and a compound that makes DNA visible under UV-light (ethidium bromide) you can see what size the fragments that you produced have. That normally gives you a good idea if the experiment ran as expected.


I did all of that and on the first try the result was no visible DNA at all. Than I changed a few parameters and tried again and saw a very thin band where I expected it and a few others where they shouldn’t be. So I changed some more parameters and on the third try I got this wonderful picture of a gel with a strong band at exactly the position where I expected it.


This made me quite happy but now I had to do something with it. I wanted to put this DNA into a yeast cell and make it integrate into the genome. This is called transformation and is quite tricky as well. I’ll explain that the next time.